Parkin (E3 Ligase) (human) (rec.) (His)
SBB-CE0055
Product group Proteins / Signaling Molecules
Overview
- SupplierSouth Bay Bio
- Product NameParkin (E3 Ligase) (human) (rec.) (His)
- Delivery Days Customer10
- CertificationResearch Use Only
- Estimated Purity>95%
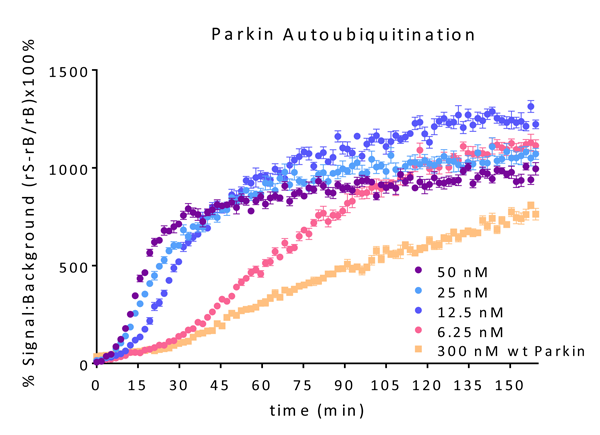
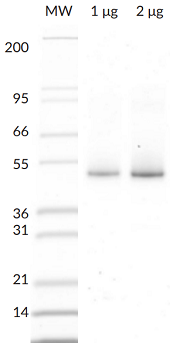
- Scientific DescriptionParkin (Parkinson juvenile disease protein 2) is a ~52kDa E3 Ligase enzyme protein, encoded by the PARK2 gene. It plays an important role in the ubiquitin-proteasome system and acts as a regulator of protein breakdown. Parkin is located in the cytoplasma until a sustained depolarization occurs after which it is translocated to the mitochondrial membrane and induces the degradation of various membrane proteins. Parkin mutation leads to the accumulation of missfolded, aggregated proteins and degenerated mitochondria. The role of these changes in the pathomechanism of neurodegenerative diseases is well-known. Parkin mutation is responsible for about 50% of familial cases and for 10 to 20% of youth cases. Present views are that the improper regulation of protein aggregation and a dysfunction of the ubiquitin-proteasome system may be the common pathway of sporadic and hereditary Parkinsons disease. In vitro, Parkin can be activated by treatment with recombinant PINK1, or addition of low concentrations of pS65-phosphoubiquitin. Parkin has been reported to generate poly-Ubiquitin chains in K6, K11, K48, and K63 linkages both in vitro and in vivo. - Protein. Human Parkin (Accession Nr. O60260) is fused at the N-terminus to a His-tag. Source: Sf9 cells. Liquid. In 50mM Tris-HCl pH 8.0, 200mM sodium chloride, 10% glycerol, 5mM TCEP. Purity: >95% (SDS-PAGE). Parkin (Parkinson juvenile disease protein 2) is a ~52kDa E3 Ligase enzyme protein, encoded by the PARK2 gene. It plays an important role in the ubiquitin-proteasome system and acts as a regulator of protein breakdown. Parkin is located in the cytoplasma until a sustained depolarization occurs after which it is translocated to the mitochondrial membrane and induces the degradation of various membrane proteins. Parkin mutation leads to the accumulation of missfolded, aggregated proteins and degenerated mitochondria. The role of these changes in the pathomechanism of neurodegenerative diseases is well-known. Parkin mutation is responsible for about 50% of familial cases and for 10 to 20% of youth cases. Present views are that the improper regulation of protein aggregation and a dysfunction of the ubiquitin-proteasome system may be the common pathway of sporadic and hereditary Parkinsons disease. In vitro, Parkin can be activated by treatment with recombinant PINK1, or addition of low concentrations of pS65-phosphoubiquitin. Parkin has been reported to generate poly-Ubiquitin chains in K6, K11, K48, and K63 linkages both in vitro and in vivo.
- Storage Instruction-80°C
- UNSPSC41116100