

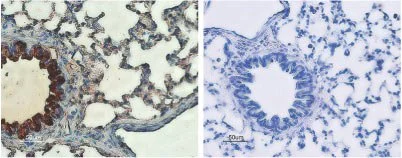
IHC-P analysis of rat lung tissue using GTX54774 CFTR antibody. Strong staining of bronchial epithelial cells (red) and lighter staining of alveolar cells (red-brown) is apparent. There is also positive staining of macrophages while smooth muscle and endothelium are negative. Counterstain of cell nuclei appears blue. A negative control is shown in the right panel.

IHC-P analysis of rat lung tissue using GTX54774 CFTR antibody. Strong staining of bronchial epithelial cells (red) and lighter staining of alveolar cells (red-brown) is apparent. There is also positive staining of macrophages while smooth muscle and endothelium are negative. Counterstain of cell nuclei appears blue. A negative control is shown in the right panel.
CFTR antibody
GTX54774
ApplicationsImmunoFluorescence, ImmunoPrecipitation, Western Blot, ImmunoCytoChemistry, ImmunoHistoChemistry, ImmunoHistoChemistry Paraffin
Product group Antibodies
ReactivityHuman, Mouse, Rat
TargetCFTR
Overview
- SupplierGeneTex
- Product NameCFTR antibody
- Delivery Days Customer7
- ApplicationsImmunoFluorescence, ImmunoPrecipitation, Western Blot, ImmunoCytoChemistry, ImmunoHistoChemistry, ImmunoHistoChemistry Paraffin
- CertificationResearch Use Only
- ClonalityPolyclonal
- Concentration0.8 mg/ml
- ConjugateUnconjugated
- Gene ID1080
- Target nameCFTR
- Target descriptionCF transmembrane conductance regulator
- Target synonymsABC35, ABCC7, CF, CFTR/MRP, MRP7, TNR-CFTR, dJ760C5.1, cystic fibrosis transmembrane conductance regulator, cAMP-dependent chloride channel, channel conductance-controlling ATPase, cystic fibrosis transmembrane conductance regulating, cystic fibrosis transmembrane conductance regulator (ATP-binding cassette sub-family C, member 7)
- HostRabbit
- IsotypeIgG
- Protein IDP13569
- Protein NameCystic fibrosis transmembrane conductance regulator
- Scientific DescriptionThis gene encodes a member of the ATP-binding cassette (ABC) transporter superfamily. The encoded protein functions as a chloride channel, making it unique among members of this protein family, and controls ion and water secretion and absorption in epithelial tissues. Channel activation is mediated by cycles of regulatory domain phosphorylation, ATP-binding by the nucleotide-binding domains, and ATP hydrolysis. Mutations in this gene cause cystic fibrosis, the most common lethal genetic disorder in populations of Northern European descent. The most frequently occurring mutation in cystic fibrosis, DeltaF508, results in impaired folding and trafficking of the encoded protein. Multiple pseudogenes have been identified in the human genome. [provided by RefSeq, Aug 2017]
- ReactivityHuman, Mouse, Rat
- Storage Instruction-20°C or -80°C,2°C to 8°C
- UNSPSC41116161
Datasheet
Related products
Product group Antibodies
Anti-CFTR AntibodyA96225
ApplicationsWestern Blot, ELISA, ImmunoHistoChemistry
ReactivityHuman, Mouse, Rat
- SizePrice
Product group Antibodies
Anti-CFTR Antibody144-66601
ApplicationsWestern Blot
ReactivityHuman
TargetCFTR
- SizePrice
Product group Antibodies
CFTR C-terminus Monoclonal AntibodyBSM-60568M
ApplicationsImmunoFluorescence, ImmunoHistoChemistry, ImmunoHistoChemistry Frozen, ImmunoHistoChemistry Paraffin
ReactivityHuman
TargetCFTR
- SizePrice
Product group Antibodies
Anti-CFTR (Phospho-Ser737) AntibodyA00028S737
ApplicationsImmunoFluorescence, Western Blot, ImmunoHistoChemistry
ReactivityHuman, Mouse, Rat
TargetCFTR
- SizePrice

Product group Antibodies
CFTR AntibodyCSB-PA001608
ApplicationsWestern Blot, ELISA, ImmunoHistoChemistry
ReactivityHuman, Mouse, Rat
TargetCFTR
- SizePrice
Product group Antibodies

Goat anti-CFTREB06694
ApplicationsELISA, ImmunoHistoChemistry
ReactivityBovine, Canine, Human, Mouse, Rat
TargetCFTR
- SizePrice
Product group Antibodies
ApplicationsImmunoPrecipitation, Western Blot, ImmunoCytoChemistry, ImmunoHistoChemistry
ReactivityPorcine
TargetCFTR
- SizePrice
Product group Antibodies

CFTR AntibodyLS-C409922
ApplicationsWestern Blot
ReactivityHuman, Mouse
TargetCFTR
- SizePrice
![IHC-P analysis of human colon carcinoma tissue using GTX22784 CFTR antibody [CF3]. Left : Primary antibody Right : Negative control without primary antibody Antigen retrieval : heat induced antigen retrieval was performed using 10mM sodium citrate (pH6.0) buffer, microwaved for 8-15 minutes Dilution : 1:200](https://www.genetex.com/upload/website/prouct_img/normal/GTX22784/GTX22784_1104_IHC-P_w_23060620_131.webp)
Product group Antibodies
CFTR antibody [CF3]GTX22784
ApplicationsFlow Cytometry, ImmunoFluorescence, ImmunoPrecipitation, Western Blot, ImmunoCytoChemistry, ImmunoHistoChemistry, ImmunoHistoChemistry Paraffin, Neutralisation/Blocking
ReactivityHuman, Mouse
TargetCFTR
- SizePrice